In a disappointment to Alzheimer’s patients and researchers, drugmaker Eli Lilly said in late November that a clinical trial of solanezumab, an experimental medication to treat the degenerative neurological condition, had failed.
The company has pressed on with tests of solanezumab, despite mixed results in earlier studies. The latest test, involving more than 2,000 patients, found the drug didn’t significantly slow cognitive decline in patients with mild dementia from Alzheimer’s.
The sad refrain is a familiar one, unfortunately.
Solanezumab is just the latest casualty in a decades-long parade of disappointing dementia drug trials. But the frustration brought by this particular failure could signal a shift in Alzheimer’s research — a shift away from targeting accumulations of so-called amyloid protein in the brain, long considered by many in the field to be the crux of Alzheimer’s pathology.
Ever since Dr. George G. Glenner’s 1984 discovery that amyloid is the main component of the plaques that riddle the Alzheimer’s-afflicted brain, it has been assumed that the protein somehow contributes to the disorder — that it jams up cellular machinery, rendering neurons unable to effectively communicate, to form new memories, to remember where the keys are.
Like many other failed medications for symptomatic Alzheimer’s, solanezumab works by attacking amyloid in the brain.
So in light of the new findings, is it finally time to let the amyloid theory go? The answer isn’t clear.
“The low magnitude of effects would lend support to the idea that it might be time to move on from amyloid,” says Weill Cornell Medical College neurologist Dr. Richard Isaacson, who wasn’t involved in the solanezumab study. “Yet though the study failed overall, there were improvements in cognition and function in treated patients.”
He points out that perhaps the tested dose wasn’t high enough or that the patients’ disease was too advanced to respond. By the time symptoms of Alzheimer’s arise, the brain is already speckled with amyloid. Two other ongoing trials should confirm whether solanezumab is more effective in patients at risk for Alzheimer’s, but who have not yet developed symptoms, he says.
Solanezumab, an antibody, works by attacking amyloid floating in cerebrospinal fluid. A different type of investigational medication, so-called BACE inhibitors, prevent amyloid formation in the first place, by neutralizing an enzyme that cuts away amyloid from a larger protein. Biogen’s aducanumab, another experimental drug that’s far along in clinical testing, binds to and clears amyloid that is already ensnared in plaques.
Earlier this year the FDA granted aducanumab fast-track status after results from a small, early-stage study suggested that it reduces amyloid plaques and slows cognitive decline in people with very early stage disease. Those people did have amyloid deposits visible with positron emission tomography imaging. At the Clinical Trials on Alzheimer’s Disease and Dementia meeting in San Diego in early December, follow-up data were presented that confirmed cognitive improvement out to two years of treatment.
“The good news is that there are a number of trials in progress with different anti-amyloid drugs in asymptomatic subjects; and that one failed drug doesn’t mean that another won’t have an effect,” says Dr. James Burke, professor of medicine and psychiatry at Duke University’s Alzheimer’s Disease Research Center. “These trials also suggest that the best chance for a significant effect on cognition is likely to be treating asymptomatic people with amyloid deposits on imaging.”
Yet, Burke adds, if these trials don’t show a significant clinical benefit, the focus on amyloid will likely end.
In any event, Weill’s Isaacson feels that researchers should be looking to other options. “I’ve never been a firm believer in the amyloid hypothesis being the be-all and end-all as to the cause of Alzheimer’s,” says Isaacson. “I think it’s much more complicated and there are probably many roads leading to the disease.”

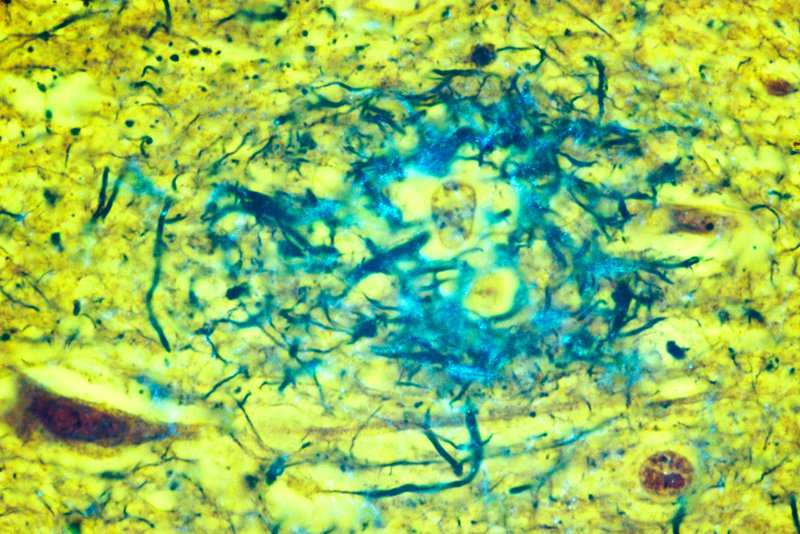
Fluorescent deconvolution micrograph of cultured glial cells expressing tau protein (in red). Glial cells are nervous system cells that provide structural support and protection for neurons (nerve cells). Accumulation of tau in brain tissue is linked with a number of neurodegenerative diseases, including Parkinson’s disease and Alzheimer’s disease.
Roger J. Bick, Kha Dinh/Mya C. Schiess / UT-Houston Medical/Science Source
One such road might be to target the tau protein, which also accumulates in tangles inside the Alzheimer’s-hindered brain. Another involves treating the inflammation that occurs with dementia, as the immune system attempts to clear clustered amyloid. Even simpler are dietary interventions. Mediterranean-like diets high in omega-3 fatty acids show particular promise in slowing cognitive decline.
As in so many other disorders, fully understanding Alzheimer’s disease might ultimately entail figuring out how our bodies interact with the trillions of microbes living in our guts, or our “microbiota.” Research in animals and humans suggest that certain combinations of these organisms may rev up the immune system in ways that contribute to dementia. A study published in July in Scientific Reports found that a long course of antibiotic treatment to alter gut flora in dementia-prone mice reduced the number and size of amyloid plaques in the brain.
Whether it’s antibiotics, probiotics or vaccines, the list of potential Alzheimer’s treatments being considered goes on.
“The bottom line is we need to take more shots on goal,” says Isaacson. “The next frontier is recognizing that there probably isn’t a one-size-fits-all approach, and that using targeted therapies based on a person’s own biology and genetics will bring the most benefit. The future of Alzheimer’s therapeutics is in precision medicine.”
Bret Stetka is a writer based in New York and an editorial director at Medscape. His work has appeared in Wired, Scientific American and on The Atlantic.com. He graduated from University of Virginia School of Medicine in 2005. He’s also on Twitter: @BretStetka.
source:www.npr.org

